MSA required format
If you choose to upload your own multiple sequence alignment, the file should have a very specific format. Pay careful attention to the details. How to construct an alignment with the desired format and a concrete example are at the end of this page. Uploading your own alignment is not recommended unless you have a very good reason.
Format details
- A plain text file (not word nor rich text format (.rtf)). Use simple text editors.
- The MSA should be in a FASTA format.
- At the top of your MSA should be two sequences.
- The sequence titles
- The 1st sequence should have one of the following titles depending on your structure input:
- The pdb_code if you used a pdb_code. For instance, >1NDD
- The file name without the suffix if you used the upload option. For instance >1NDD_uploaded if the uploaded structure file had the name 1NDD_uploaded.pdb
- The 2nd sequence should have the same name as the 1st sequence with the suffix _pdb. For instance, 1NDD_pdb
- The 1st sequence should have one of the following titles depending on your structure input:
- The amino acid sequences should be as follow:
- The 1st sequence should be the full sequence.
- If you entered a pdb_code, you can find and copy the full sequence in the RCSB web by clicking Display files and then FASTA sequence. Example
- If you uploaded your own files, the amino acid sequence should be the same as in your uploaded sequence file.
- The 2nd sequence should be the amino acid sequence in the PDB. It may be identical to the 1st sequence but will differ in cases of lack of density. One way to get the PDB sequence is by extracting it using PyMOL. Another way is loading to RCSB sequence webpage. Example
By examining the schematic representation of the amino acids and the secondary structure content shown above the letters, you can identify amino acids that were not crystallized (i.e., there is no secondary structure description above them). Then, you can copy the full sequence which you already obtained and simply delete all the amino acids that were not crystallized. For instance, for 1NDD, the last two amino acids (GG) should appear in the 1st (full) sequence but not in the 2nd (PDB) sequence.
- The 1st sequence should be the full sequence.
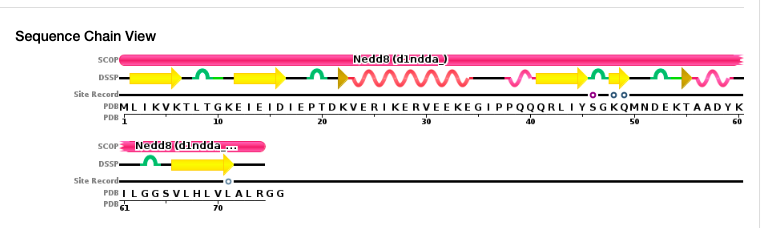
How to construct an alignment with the required format
- Start by getting the 1st and 2nd sequences as described above.
- Copy these to the list of homologues that you already have (preferably before they were aligned), and give them names as described above.
- Obtain a multiple sequence alignment using MUSCLE or other softwares.
**if you already have a multiple alignment and do not have the homologues in a file version that is not aligned (i.e., without hyphens), add manually the 1st and 2nd sequences to this file. Then, manually add hyphens to make these two sequences match the rest of the sequences. THIS IS LESS RECOMMENDED AS IT IS ERROR PRONE. - Open the derived multiple alignment. The two sequences may not be the firsts after step 3. Identify the sequences by their name, cut and paste them back at the top of the file.
- If the PDB (2nd sequence) has lack of density, carefully examine the multiple alignment results to see that the indels between your two sequences were assigned correctly. SOMETIMES MUSCLE AND OTHERS ARE WRONG :(
An example of how the first two sequences should look, given on the PDB entry 1NDD
>1NDD
---MLIKVK-----------TLT------G--------------------EI-DIEPTD-
------------------K-----------------------------------------
---VERIKER---------V----------------------------------EEK-E-
-GIPPQ-------------------------------QQ---------------------
---------R-------LIYS--------GKQM---------------------------
---NDEKT-AADYK---I--LGG-----SVLHLVLALRGG
>1NDD_pdb
---MLIKVK-----------TLT------GK----------------E-IEI-DIEPTD-
------------------K-----------------------------------------
---VERIKER---------V----------------------------------EEK-E-
-GIPPQ-------------------------------QQ---------------------
---------R-------LIYS--------GKQM---------------------------
---NDEKT-AADYK---I--LGG-----SVLHLVLALR--
>TGJ86544_61_100 | a third homologue from the full alignment
---MMIKVR-----------TLT------GK----------------L-IEF-DVEPTD-
------------------T-----------------------------------------
---IEDVKKR---------V----------------------------------EEK-E-
-GILPA-------------------------------QQ---------------------
---------R-------LIYA--------GKQM---------------------------
---HDEVK-LGEGG---V--VAD-----ATLHLVLTLRGG